How To Take 3-D Data
written by Hetal Patel & edited by Eric Weeks
Lab Home --
People --
Experimental facilities --
Publications --
Experimental pictures
-- Links
Getting Data using Epoxy
|
First, start up Intervision as normal. Using whatever you need for Brightness, Contrast, Slit settings, Barrier Filter, Laser Light, and Laser Intensity, focus your slide as normal. Also, make sure that your zoom level is zoom: 1.00. Once this is done, click on Intervision Acquisition Tool, pull down the Protocols Menu, move down to Load, and over to ZSeries. This will pop up a new window called ZSeries.
Once there, click on Spacing in the upper left and change the 0.500 to 0.2.
Next, change the Resolution from Medium to Low. If your particles are crappy, you can use jump average to your heart's content. (I prefer live though).
|
Crucial parameters:
- Zoom = 1.0
- Z-spacing = 0.2 microns
- Resolution = low
|
|
Once this is done, click on Motor at the top right, and move down to Piezo. This will open up the Piezo popup box. Adjust the Piezo so that it is where you want your 3-D data to start. Once you have the correct position, click back on Z Series and click on the Start
button at the top right. Then, click back on the piezo and adjust it until it is at the position that you want your 3-D data to end. Once you have that, click back on Z Series and click on End (also top right). Typical values are start at 50 microns, end at 60 microns (with 0 microns being the location of the coverslip). You can now close the Piezo popup if you so desire.
Now, click on the Time Lapse button on the Z Series box. This will open up a new popup. At the top, Stacks is how many individual data files you want. I normally choose 30, but if you want more data, feel free to choose 50 or 100 or whatever. Then, click on Seconds on the 3rd line. This will tell the Piezo how much time it has to move through the sample and then back to the top before starting over. Normally, I have found that if you use a difference of about 25 microns from start to finish, it will need about 30 seconds to properly go through the sample in time. Feel free to experiment with this as you so choose. You can tell whether you've picked an appropriate time by actually taking data (Don't do so now). I will explain this in a bit.
Next, still in the Time Lapse Setup popup box, click on Base Name and enter the name that you want the files to have. All files will start with this name and end in 0000 through 00X where X equals (# of stacks)-1. There is also an option for Hard Shutter. When this is depressed (yellow), it means that in between stacks, it will turn the shutter off to save laser power. Normally this is good for long samples where the interval period is minutes or even hours, so that bleaching of the sample does not occur. Again, feel free to experiment with this option too.
Now, press the Close button. To take data, click on the Record dot in the Intervision Acquisition Tool. To see whether or not you picked a good interval time, wait until the first data is taken. After it is taken, it will say Saving at the top right after Status. After Saving, it should give you a countdown of numbers, normally from 10 down to 0. When it reaches 0, it starts taking data again. You need this countdown to start at LEAST from 2. If you do not see this countdown, you need to stop taking data and click back on Time Series and increase the interval time. Likewise, if the numbers are too high (i.e. starting from 60 and counting down), you can reduce the interval time so that your sample is done in an ideal amount of time. In a fast moving sample, having the numbers too high makes it hard for the tracking program to accurately relate particles in various frames, so be wary. Again, this depends on what exactly you want done and whether you want to take a long data set or a short one. When data taking is done, the Status will say Live and you can close intervision, or take more data.
Pretracking Data
Transfer your data from Epoxy to Spicysquid using FTP:
spicysquid% ftp epoxy
ftp> ls
ftp> prompt
ftp> mget yourfiles*
ftp> bye
The mget command grabs multiple files. The prompt command tells FTP not to ask you are you sure? for each of these multiple files.
Open up IDL in the directory that you moved the files to. To pre-track your data, type in pt3d,'base_name_of_file*' where the base_name_of_file is what you entered in when you took the data.
This will take a nice amount of time depending on how much
data you had and how many microns you were analyzing, so go take
a break and have some lunch or a beer while Spicysquid does some work. When it's done, type in pta=catptdata('pt.base_name_of_file*'). This will combine all your pretrack data into a single file so that you can analyze it. Once that is done, write out that file using write_gdf,pta,'ptbase_name_of_file' to write out your file. Next, type in help,pta. This will bring up something that tells you the dimensions of your data in a matrix form. Normally there are two numbers, the first number being a single whole number and the second number being a 5 to 7 digit whole number. Basically, just look for any abnormalities.
To look at your data, you need to type in plotp,pta.
This will open up an IDL screen that plots each and every
single one of your data points. Again, look for any
abnormalities--weird stuff that you don't think should be
there. Now, type in plot,pta(3,*),pta(4,*),ps=3
this will give you a general plot of your data so
that you can crop it. Basically, you need to get
the main body of the data and remove outliers so
that you won't have problems later on. Use this command
ptb=eclip(pta,[3,(left_edge),(right_edge)],[4,(bottom_edge),(top_edge)])
where the y-axis is the 4 category and the
x-axis is the 3 category to crop your data.
For instance, one of them could look like this:
ptb=eclip(pta,[3,0,4e4],[4,4,12]) This would crop
everything except the region between 0 and 4e4 on the x-axis
and between 4 and 12 on the y-axis leaving you a nice little
bubble. There's a description of this clipping procedure at the
particle tracking tutorial web page
After you have done this, type in ptb(0:1,*)=ptb(0:1,*)*0.2352 and then ptb(2,*)=ptb(2,*)*0.2 to normalize your data. If you are wondering where the two numbers came from, the 0.2352 came from when you first entered in your noran data, it is the x and y
calibration (microns per pixel). (So your data probably came out like (0.2352, 0.2352, 0.2). Likewise, the 0.2 came from the final part of the original entering. It is basically the number for the SPACING that you typed in when you originally took the data.)
Finding the pair correlation function
|
Once that is done, write your file out using write_gdf,ptb,'ptb_base_name_of_file'. Now, type in emgrwrap,'ptb*',/special,/pretrack. This is Eric's program to view the pair-correlation function of your data. It will take a short amount of time and should go through all the data files and create one large file that starts with a gr so that you can easily view it. To view this gr file, type in gr=read_gdf('gr.pt*') and then plot,gr(0,*),gr(1,*) and this should bring up a graph that looks identical to the final one that popped up at the end of the pair correlation; see the picture at right. The x-axis is in microns; the first peak indicates the most probable separation for nearest neighbors, and the second smaller peak is the 2nd-nearest-neighbors, etc.
|
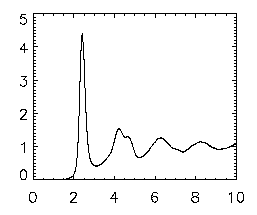
|
If you're interested in looking at the differences in two different pair correlation plots, you can use the oplot command to compare two. First, enter in your second gr data by using gr2=read_gdf('gr.pt_name_of_file') Note: if you do DO an overplot, make sure you change the asterisk (*) in the gr=read_gdf command above to the base name of the file that you want as the first gr and use the base name of the file that you want as the SECOND gr in the gr2=read_gdf command. To overplot, simply plot the first gr as normal and then type oplot,gr2(0,*),gr2(1,*),lines=1 where 1 can be any whole number up through 10.
Next, if you want to see where the peak positions are and the height and so on, use the following command. grwrap3,'gr.pt*' and this will show you the peak and heights and other information of the data on your pair correlation. Again, if you had more than one gr, make sure you use the appropriate name of the file instead of
gr.pt*.
Finding the volume fraction
Start by making a ptb file as described above. The main important points are that the data need to be calibrated into microns, and you need to have clipped out any bad particles. Then run two commands:
- volwrap,'ptb*'
- volwrap2,'vol*'
The first command makes a bunch of files with the voronoi volume for each particle. The second command figures out the average volume per particle and prints it to the screen. At the moment, the diameter of the rhodamine-dyed PMMA particles (ASM57) in a cyclohexylbromide/decalin mixture is believed to be 2.33 microns, plus or minus 0.01 microns. This applies definitely to Eric's slides 310-330.
Using color in graphs
|
If you wish to show colored lines in IDL, use the following commands before you plot your graph:
- device,decompose=0
- tek_color
After you plot your first line for your graph, add ,color=X (where X is any whole number up to 30) to the end of your oplot command to make the line that color.
|
Click on picture for an enlargement
|
Any comments, let Eric Weeks <weeks(at)physics.emory.edu> know.
